
anilvaishnavi@gmail.com
Recent Articles
Health problems related to food & water borne infections are very common in summer and acute gastroenterites is one of them. The common symptoms are vomiting, nausea, diarrhoea, stomach cramps, and dizziness. Diarrhoea & vomiting is usually because of unsafe food & water, poor sanitation, lack of hygiene and ignorance. Children are more prone to vomiting and diarrhoea, and are more likely than adults to die from diarrhoea, because they become dehydrated more quickly, as compared to adults.
Diarrhoea is also major cause of child malnutrition. Every minute, one child dies in India because of diarrhoea, i.e diarrhoea kills as many as 5 lacks children under five years, every year. According to Dr. Anil Vaishnavi, Child Specialist & Neonatologist at Batra Hospital New Delhi, many of these deaths can be prevented if WHO recommended ORS is given promptly & after every stool. He also adds, to avoid diarrhoea,& vomiting, simple preventive measures should be taken like, washing hands before eating, cutting of nails regularly, eating fresh and healthy food and drinking clean and safe water. In case of severe dehydration, the child must be admitted to a hospital,” says Dr. Anil Vaishnavi.
Many different sorts of germs can cause diarrhoea & vomiting. Good food hygiene standards in the food industry and at home are vital to prevent food borne diseases. The main causes of food poisoning and food borne diseases are: Preparing foods too far in advance, Not cooking foods properly, Not defrosting foods correctly, Storing foods incorrectly so that bacteria can grow quickly, Contamination from people handling foods due to poor personal hygiene. Some of the preventive measures are:
| Wash your hands after using the toilet and before handling food. | |
| Do not use the same towel or face cloth as someone who is suffering with food borne illness. | |
| Clear up soiling accidents straightaway, wash with hot soapy water and disinfect with a disinfectant or bleach. With bleaches and disinfectants always follow the instructions on the packaging. | |
| Disinfect door and toilet handles, taps and the toilet seat after use and disinfect the toilet bowl frequently. | |
| Drink plenty of fluids while you are ill to prevent dehydration |
Article Published in Gurgaon, News of the Week on 24/06/2006
SHOCK IN CHILDREN:
Shock is one of life threatening conditions faced by paediatric in critical care settings. In our PICU one in every fourth admitted to level III care has symptoms of shock.
Shock can be defined as acute circulatory failure with inadequate or inappropriate tissue perfusion resulting in generalized cellular hypoxia. Shock is not a problem of blood pressure or blood volume, whatever the underlying cause it is always a problem of inadequate cellular sustenance. All forms of shock can result in impaired function of vital organs such as brain (depressed mental status) & kidneys (Low urine output, in effective filtering) & untreated this leads to metabolic acidosis & organ dysfunction & death.
Classification of Shock:-
(A) Hypovolemic shock
(B) Cardiogenic shock
(C) Obstructive shock
(D) Distributive shock
(E) Dissociative
Phases of shock:-
(a) Compensated Shock:- Vital organs function is maintained primarily by intrinsic compensatory mechanisms. Early recognition of compensated shock is critical to effective treatment & good out come.
(b) Uncompensated State:- Microvascular perfusion becomes marginal despite compensatory mechanisms. Cellular function deteriorates & disturbance in function becomes evident in all organ systems.
(c) Terminal or Irreversible Shock:- Implies damage to key organs, to such magnitude that death occurs even if therapy returns, Cardio vascular parameters to normal levels.
Warning signs:-
| Marked Tachycardia | |
| Weakening central pulses | |
| Cold distal extremities with very prolonged capillary refill | |
| Narrowing pulse pressure | |
| Altered mental status | |
| Hypotension (Late findings) |
Two major priorities in treatment of shock are:
a) Rapid assessment of patients disease process
b) Achievement of cardiopulmonary stability
Fundamentals of shock management:-
| Position the child |
|
| Optimize arterial oxygen content | |
| Support ventilation as indicated (Invasive or noninvasive) | |
| Establish vascular access |
|
| Begin fluid resuscitation | |
| Start monitoring |
|
| Perform frequent reassessment |
|
| Conduct ancillary studies |
|
| Administer pharmacologic support- See Table-2: Pharmacologic Agents used in Treatment of shock. |
|
| Obtain subspecialty consultation |
Table 2: Pharmacologic Agents Used in the Treatment of shock
Class |
Drug |
Effect |
| Inotropes | Dopamine Epinephrine Dobutamine |
|
| Phosphodiesterase Inhibitors (indicators) |
Milrinone Inamrinone |
|
| Vasodilators | Nitroglycerin Nitroprusside |
|
| Vasopressors (vasoconstrictors) |
Epinephrine Norepinephrine Dopamine Vasopressin |
|
Monitoring required in patients during shock for better out come:
(A) Invasive homodynamic monitoring, which includes
| Central Venous Pressure (CVP) | Mean arterial pressure (MAP) |
| Mixed venous oxygen saturation (Snvo2) or Central venous oxygen saturation (SVo2) |
(B) Fluid Resuscitation
| Type of shock | Volume of fluid | Rate of Delivery |
| Hypovolemic shock (non DKA) Distributive shock Obstructive shock |
20 ml / kg bolus (Repeat PRN) |
Deliver rapidly (Over 5-10 min.) |
| Cardiogenic shock (non poisonings) |
5-10 / Kg bolus (Repeat PRN) |
Deliver more slowly (Over 10-20 min) |
| Diabetic Ketoacedosis (DKA) |
10-20 ml / kg | Deliver over 1 hour |
| Poisonings (e.g.) Calcium channel blocker or ß- adrenergic blocker |
5-10 ml / kg (repeat PRN) |
Deliver more slowly (over 10-20 min) |
(C) Glucose Monitoring
In all critically ill & injured children, hypoglycemia should be treated effectively & enthusiastically as it leads to brain injury. While some seriously ill or injured children can have hyperglycemia, it is recommended to correct hyperglycemia in high risk groups, such as brain injured children.
Untreated shock is universally lethal, but with proper recognition, monitoring & treatment, the morbidity & mortality can be reduced significantly.
Article Published in Niramayam Vol 2010, Issue 1
PERSISTENT SYMPTOMS
APPROACH TO PERSISTENT CHOLESTATIC JAUNDICE
The approach to persistent conjugated hyperbilirubinemia is directed by the age at presentation. Two categories can be defined; the cholestatic baby (< 1year) and the older child ( >1 year). Although there is an overlap in the disorders producing persistent jaundice in these two categories, the approach is the distinctive.The approach is directed by the clinical features and employs many different investigational methods including biochemistry, hematology, microbiology , radiology, nuclear medicine and histology. As the clinical presentation of many diseases is similar, it is appropriate to perform a variety of first line tests, proceeding to more complex investigations only when indicated.
Approach in the cholestatic baby ( < 1 year )
If jaundice has presented in the first few weeks of life, the main differential diagnosis is between extrahepatic biliary atresia (EHBA), neonatal hepatitis or other disorders involving the biliary tree ( Table 1 ). A number of clinical clues will be evident from the clinical or family history and physical examination and the series of investigations will be directed by this information. Affected infants have icterus, dark urine, light or acholic stools and hepatomegaly, all reflecting decreased bile flow resulting from either liver cell injury or bile duct obstruction. Babies with EHBA usually are born at term, are of average weight and initially thrive well. In contrast babies with intrauterine infections are commonly born prematurely, are small for date and feed poorly. The visual inspection of the stool is of paramount importance. The presence of persistent pale or acholice stools is a pointer towards EHBA and warrents an urgent workup. The presence of abnormal facies (broad forehead, widely spaced eyes and pointed chin), systolic murmur of pullmonic stenosis and posterior empryotoxon would suggest Alagille’s syndrome. Babies with intrauterine infections may have dysimorphic features with characteristic eye findings.
Although standard liver function tests are unlikely to be helpful in the differential diagnosis between biliary obstruction and neonatal hepatitis, gamma glutamyl transferase and alkaline phosphatase show significant derangement in biliary obstruction. The chronicity of the liver disease may be implied by low albumin concentration and the prolonged coagulation time, which are unresponsive to vitamin K therapy. Poor hepatic function at birth suggests that the disease process has existed in utero and may be either an inborn error of metabolism or an infection. At most neonatal liver diseases present in similar way, the next step is the prompt recognition of any specific or treatable primary cause of cholestasis such as endocrinopathy (hypothyroidism or panhypopituitarism). Nutritional hepatotoxicity caused
Table 1. Differential diagnosis of neonatal cholestasis
|
Table 2. Workup for neonatal cholestasis
|
Specific metabolic diseases such as lactosemia or tyrosinemia type I. It is casual to perform a series of first line tests in order to exclude the known causes of intraurine infection and certain inborn errors of metabolism and to establish the patency of ktraheptic biliary tree (figure 1). More specialized tests are only performed if clinical suspicion is high or the first line tests suggest an inborn error of metabolism (table 2).
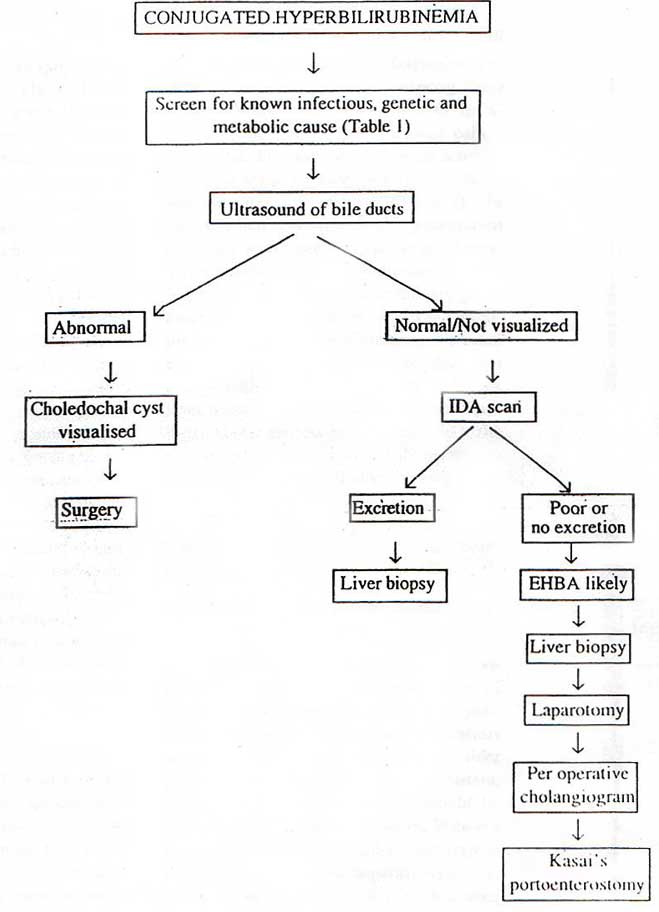
It is important to exclude extrahepatic biliary atresia or a choledochal cyst at an early age as corrective or palliative surgery can be performed early. Abdominal ultra-sound is a useful investigation, which may demonstrate a choledochal cyst or an absent gall bladder (suggestive of EHBA). If the fasting ultrasound is normal or equivocal it is necessary to perform a radioisotope scan using technetium IDA (imminodiacetic acid) to demostrate hepatic uptake of isotope and excretion into the bowel. In EHBA, hepatocyte function is intact and uptake of the agent is unimpaired but excretion into the intestine is absent. In patients with neonatal hepatitis uptake is sluggish, but excretion into the biliary tract and intensive eventually occurs. Oral administration of Phenobarbital (5 mg / kg / day) for 5 days prior to the study enhances biliary exception of the isotope in patients with neonatal hepatitis.
Liver biopsy provides the most reliable discriminatory evidence and may differentiate between intra and extra hepatic disorders, but there is considerable overlap in the patho-logical features. In EHBA, there is bile ductular proliferation, the presence of bile plugs and fibrosis with basic hepatic lobular architecture intact. In neonatal hepatitis, ther is severe, diffuse, hepatocellular disease with distortion of lobular architecture, marked in filtration with inflammatory cells and focal hepatocellular necrosis. While the bile ductules show little alteration. Giant cell transformation is found in infants with either condition and has no diagnostic specificity. Histological changes similar to those in idiopathic neonatal hepatitis occur in a variety of diseases including alpha-1 antitrypsin deficiency, galactosemia and various forms of intrahepatic cholestasis. Although, paucity of intrahepatic bile ductules may reveal a more characteristic pattern.
In extrahepatic biliary obstruction it may be necessary to perform a laparotomy and an operative cholangiogram to outline the extrahepatic biliary tree before diagnosis. The role of magnetic resonance cholangiopancreatography remains to be defined in babies with EHBA.
Hypothyroidism is usually associated with unconjugated hyperbilirubinemia but it may exacerbate underlying hepatitis. T4 and TSH should therefore, be measured. A fasting cortisol level should be measured to exclude panhypopituitarism due to septo-optic dysplasia. Chromosomal studies should be performed because of the association between neonatal hepatitis and trisomy 13 and 18. Sweat test should be done to exclude cystic fibrosis which usually presents with neonatal hepatitis.
The commonest inborn error of metabolism to present with neonatal cholestasis is alpha-1- antitrypsin deficiency which can be diagnosed by establishing a reduced serum level (<0.8 g/L) of alpha-1- antitrypsin. In the presence of a low level, a phenotype analysis by isoclectric focusing is warranted to confirm the presence of phenotype PiZZ. Liver histology will confirm storage of PAS positive granules in the endoplasmic reticulum. While inborn errors of carbohydrate metabolism will be evident by the detection.
Figure1: Approach to neonatal cholestasis
of reducing sugars in urine or a hypoglycemia (<30mg/dl) and or a raised plasma lactate(>3.0 mmol/l) babies with severe liver disease may have gross galactosuria. Estimation of the enzyme galactose-I- phosphate uridyl transferase in red blood cells is required to confirm the diagnosis of galactosemia. Hereditary fructose intolerance present later in infancy when sucrose has been introduced to the diet and is confirmed by estimating fructose 1-6 diphosphate aldolase in liver or intestinal biopsies. Glycogen storage disease does not present with neonatal cholestasis but with hepatomegaly and some variants present with hypoglycemia and a raised plasma lactate. Tyrosinemia type I is the commonest disorder of amino acid metabolism and is suggested by finding elevated levels of tyrosine and methionine in plasma and urine. However, non specific increase can occur in severe liver disease from any cause, it is therefore important to measure the urinary succinyl-acetone in a suspected case of tyrosinemia type I. The diagnosis is confirmed by demonstrating a deficiency of fumaryl acetoacetate in leukocytes or cultured skin fibroblasts. An elevated alpha fetoprotein is in addition suggestive evidence of tyrosinemia and may indicate early malignant change in the liver, which can be confirmed by abdominal ultrasound, CT or liver histology. Niemann-Pick disease type C is the commonest lipid storage disease to present with neonatal hepatitis. There is no screening test, but the diagnosis must be excluded by looking for the characteristic storage cells in liver, in bone marrow and in ganglion cells on rectal biopsy. In Gaucher disease glucosylceramide is stored in the reticuloendothelial system and the enzyme determination can be performed on white blood cells or cultured skin fibroblasts. Neurological problems in babies with neonatal liver disease may be primary (e.g. Zellweger syndrome) or secondary to unrecognized hypoglycemia, hyperammonemia or intracranial hemorrhage. Most of these babies will not be cholestatic and if they are actually ill one needs to exclude urea cycle defects and fatty oxidation defects (Figure 2). In neonatal hemochromatosis the presentation is usually with acute liver failure in infancy. The diagnosis is made by finding of high ferritin and iron deposition in salivary gland biopsy. A liver biopsy usually not possible because of deranged clotting. Familial erythrophagocytic lymphohistiocytosis can present with cholestasis. A high triglyceride level, a low fibrinogen and characteristic findings on bone marrow examination lead to the diagnosis. In patients with persisting cholestasis in whom all other investigations for inborn errors of metabolism are negative, urine should be screened for inborn errors of bileacid biosynthesis using fast atom bombardment mass spectroscopy (FAB-MS)
Approach in the Older Child (>1 year)
In children, hepatitis secondary to viral infection accounts for most cases of cholestasis with onset after the first year. Conditions producing neonatal cholestasis may also cause chronic cholestasis in older patients. An older child with persistent, conjugated hyperbilirubinemia, should be evaluated for chronic viral hepatitis, Wilson’s disease (older>3 years) alpha-I- anti trypsin deficiency autoimmune hepatitis type I and II, the syndromes of intrahepatic bile duct paucity, primary sclerosing cholangitis and liver disease associated with inflammatory bowel disease(Table 3). Other causes include abdominal tumours, enlarged lymph nodes, obstruction caused by cholelithiasis, hepatic inflammation resulting from drug ingestion (chlorpromazine, nitrofurantion, chemotherapy and total parenteral nutrition). Standard biochemical tests may show raised transaminases 4 to 10 times upper limit of normal or evidence of poor hepatocellular function (low albumin and abnormal coagulation).
Figure2: Differential diagnosis of metabolic liver disease in infants
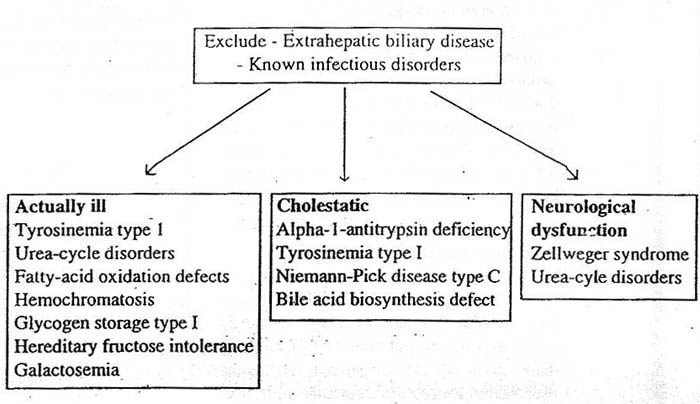
Table 3. Different Diagnisis of cholestasis in the older child
| Chronic persistent / active hepatitis Hepatitis B Hepatitis C Autoimmune hepatitis Type-I Type-II Primary sclerosing cholangitis Drugs Wilson’s disease (>3 years) AlphaI- antitrypsin deficiency Cystic Fibrosis Conditions presenting in infancy Paucity of intrahepatic bile ducts Progressive familial intrahepatic cholestasis |
Viral etiology must be excluded at all times (table4). Autoimmune liver disease is present in both sex, however the incidence is higher in girls (3:1). Non organ specific auto antibodies (SMA, anti LKM antibodies) may be demonstrated in 70% of children and there is always an increase in IgG and a positive ANA14 . In primary sclerosing cholangitis characteristic beading and narrowing is seen on endoscopic retrograde cholangio pancreatography. Wilson's disease may present with any form of liver disease and it must be considered in children over 3 years of age. Classically the diagnosis is established by detecting a reduced serum copper (< 10umol/l ), ceruloplasmin (<200mg/l ) and excess urine copper (>1.0umol/24hr). Approximately 24% of children presenting with hepatic disease may have normal or borderline ceruloplasmin but all should have elevated urinary copper excreation, especially after penicillamine challenge test (20mg/kg). Also there will be increased hepatic copper on liver histology (>250mg/g of dry weight) which is higher than the amount detected in chronic cholestasis. In equivocal cases radioactive copper studies either with 64cu or 67 will demonstrate reduced incorporation into ceruloplasmin compared to normal. In neurological presentation of Wilson disease, KF ring is always diagnostic and is present in all cases.
An abdominal ultrasound may show an enlarged spleen suggesting cirrhosis and portal hypertension from any cause. The presence of oesophageal varices and gastropathy on endoscopy confirm portal hypertension. Radiographs of the wrist, which may show osteopenia or rickets will be evidence for chronic liver disease. Severe rickets suggest a renal tubular disorder that is usually secondary to an inborn error of metabolosm e.g. Wilson’s disease; hereditary fructose intolerance tyrosinemia type I. A liver biopsy is invaluable for the purpose of diagnosis, prognosis and in some cases assessing response to therapy (hepatitis B and C).
Table 4. Workup for cholestasis in the older child
| I) History and Physical examination size and consistenancy of liver and spleen presence of stigmata of chronic liver disease, skin, eye, joint abnormalities.
II) Investigations Hepatitis Bs Ag If + ve then Hepatitis Be Ag Hepatitis B core IgM Hepatitis B Core IgG Hepatitis B DNA(qualitative) Hepatitis C Antibody (3rd generation assay) If + ve then Hepatitis C RNA Cooper Cerulopasmin 24 hour urinary copper excretion (pre and post penicillamine) ANA SMA LKM antibodies Immunoglobulins C3 and C4 Alpha-I antitrypsin level Aplha –I antitrypsin phenotype Ultasonography Endoscopy Liver biopsy Endoscopic retrograde cholangiopancreatography |
Article Published in Indian Journal of Practical Paediatrics vol 1 No. 3, Oct-Dec1999

Article Published in HT shoppers , 7th April 2011